《薬事申請のキホン》CTDって何?必須基礎知識を要点解説

医薬品メーカー等のCMC部門に携わる方であれば、一度は「CTD」という単語を耳にしたことがあるのではないでしょうか?
今回は、CTDにあまり馴染みがない方に向けて、CTDの基本知識を簡単にご紹介します。
CTDに馴染みがない方はもちろん、CTDの取扱い経験がある方も、知識のおさらいとして、ぜひ最後まで読んでみてください。
1.CTDとは?
CTDとは、医薬品を承認申請する際に添付する資料について定めたものです。
審査にかかる負担を減らし、承認審査の迅速化を図るため、記載事項だけでなく、書式やフォントサイズまで、細かく規定されています。
申請資料の配列を含む資料様式が示されていますが、承認取得に必要な試験項目を示すものではありません。
CTDの正式名称は”CommonTechnicalDocument“(共通技術文書)です。
医薬品申請に必要な申請資料の書式を国際的に共通化し、他国間申請の簡便化を図るため、2000年に日米EU医薬品規制調和国際会議(ICH)で合意されました。日本では、2003年から先発医薬品の承認申請書類に、CTDの適用が義務化されています。
CTDは、2022年9月現在、日本・アメリカ・EU・イギリスで共通して用いられる医薬品の承認申請基準です。
また、先発医薬品だけでなく、後発医薬品にも適応されています。
(1)CTDの適応範囲
CTDは、主に、バイオテクノロジー応用医薬品を含む、新有効成分含有医薬品の承認申請で必要な添付資料に適応されます。
また、薬生審査発0311第3号通知「医療用医薬品の承認申請の際に添付すべき資料の取扱いについて」において、承認審査の効率化を図るため、後発医薬品の薬事申請においてもCTDの適応が義務化されました。
なお、薬事申請における添付資料の構成について、第2部の非臨床研究結果概要、及び臨床研究結果概要においては、必要に応じて資料様式の変更が認められています。
(2)CTDの一般原則
CTDでは、審査員が効率的に内容を把握し、審査を迅速に行うため、申請資料を下記の通り規定しています。
- 曖昧さを排し、わかりやすく記載すること
- A4(日本・EU)、もしくは8.5×11インチ(米国)サイズの紙に余裕を持って印刷できるよう、余白を残して作成すること
- 左綴じをした際に情報が隠れないよう、十分な余白をもって作成すること
- 本文・表は、複写後も十分に読みやすいフォントサイズを用いて作成すること
(本文はTimesNewRoman12ポイント(英語)、もしくはMS明朝10.5ポイント(日本語)が望ましい) - 全ページにページ番号を記載すること
- 資料内で用いる頭文字・略語は角モジュールの冒頭で定義すること
- 参考文献の引用は「生物医学雑誌への投稿のための統一規定(Uniform Requirements for Manuscripts Submitted to Biomedical Journals)」の最新版に従うこと
薬事申請を行う際には、速やかに承認を得るためにも、上記原則に則したCTDの作成を心がけましょう。
2.CTDの構成
CTDの構成の概念図は、下図1の通りです。
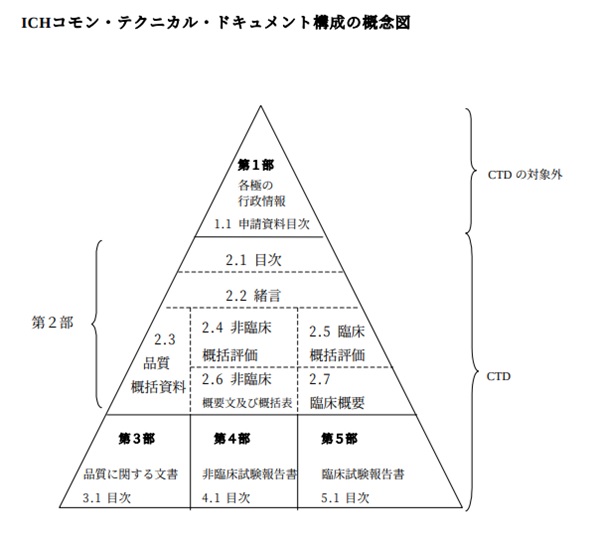
【図1 ICHコモン・テクニカル・ドキュメント構成の概念図 ※引用[1](別紙1)】
また、各部の構成の詳細については以下の通りです。
(1)第1部(Module1):申請書等行政情報及び添付文書に関する情報
第1部は、申請地域に特異的な文書が該当し、CTDに含まれません。
また、各地域の規制当局によってフォーマットが定められます。
| 第1部(Module1):申請書等行政情報及び添付文書に関する情報の構成 |
| 1.1 第1部(モジュール1)の目次 1.2 各地域に特異的な文書(申請書、添付文書(案)など) |
(2)第2部(Module2):CTDの概要(サマリー)
第2部からCTDの対象となり、下記項目を、それぞれ規定されたページ数以内で記載します。
- 目次
- 緒言
- 品質研究の概括資料
- 非臨床研究の概括評価・資料
- 臨床研究の概括評価・資料
「品質研究の概括資料」「非臨床研究の概括評価・資料」「臨床研究の概括評価・資料」の構成は、それぞれ「CTD−品質に関する文書の作成要領に関するガイドライン(M4Q)」「CTD−非臨床に関する文書の作成要領に関するガイドライン(M4S)」「CTD−臨床に関する文書の作成要領に関するガイドライン(M4E)」の規定に従います。
| 第2部(Module2):CTDの概要(サマリー)の構成 |
| 2.1 CTD全体の目次 2.2 緒言 2.3 品質に関する概括資料 2.4 非臨床に関する概括評価 2.5 臨床に関する概括評価 2.6 非臨床概要の概要文・概要表 2.6.1薬理の概要文・概要表 2.6.2薬物動態の概要文・概要表 2.6.3毒性の概要文・概要表 2.7 臨床概要 2.7.1生物薬剤学及び関連する分析法の概要 2.7.2臨床薬理の概要 2.7.3臨床的有効性の概要 2.7.4臨床的安全性の概要 2.7.5個々の試験のまとめ |
(3)第3部(Module3):品質に関する文書
第3部は、医薬品の品質に関する試験情報、製造工程プロセスのデータなどを、M4Qに従い記載します。
申請資料全体の基礎データともいえるでしょう。
また、製造工程の開発を含む製剤開発の記述は、充実したデータ記載が要求されており、特に重要な部分ともいわれています。
第3部は特に煩雑になりがちな項目を含むため、図表をうまく活用し、簡潔にまとめることがポイントです。
| 第3部(Module3):品質に関する文書の構成 |
| 1.1 第1部(モジュール1)の目次 3.1 目次 3.2 データ又は報告書 3.3 参考文献 |
(4)第4部(Module4):非臨床試験報告書
第4部は、非臨床試験結果に関する報告書を、M4S記載の順序に従い添付します。
また、個別データは、試験報告書中、もしくは試験報告書に付表として添付することも可能です。
| 第4部(Module4):非臨床試験報告書の構成 |
| 1.1 第1部(モジュール1)の目次 4.1 目次 4.2 試験報告書 4.3 参考文献 |
(5)第5部(Module5):臨床試験報告書
第5部は、臨床試験結果に関する報告書をM4Eの記載の順序に従い添付します。
ICHの原則に基づき、試験報告書に必要なすべてのセクションとサブセクションが含まれるよう規定されています。
| 第5部(Module5):臨床試験報告書の構成 |
| 1.1 第1部(モジュール1)の目次 5.1 臨床試験報告書及び関連情報の目次 5.2 臨床試験一覧表 5.3 臨床試験報告書及び関連情報 5.4 参考文献 |
3.迅速に承認を得るためのポイント
開発が競合する医薬品開発では、類似効果を持つ他社の医薬品との競争で勝ち抜くために、CTDを適切に作成し、最短期間で申請承認を得ることが重要です。
申請資料の不備によって、承認期間が長期化することがないよう心がけましょう。
わかりやすく、簡潔にまとめることが要求されるCTD資料においては、うまく図表を用いることも重要なポイントです。CTD資料の作成に不安がある場合は、薬事申請資料作成のプロへ相談するとよいでしょう。
また、医薬品開発に携わる方々に向けた、薬事申請に関する注意事項や、効率的に承認を得るコツを紹介するセミナーも定期的に開催されています。興味がある方は、ぜひ調べてみてください。
(アイアール技術者教育研究所 S・N)
≪引用文献、参考文献≫
- [1]ICH-M4CTD(コモン・テクニカル・ドキュメント)|独立行政法人医薬品医療機器総合機構
※別紙1(コモン・テクニカル・ドキュメント(CTD)の構成) - [2]申請区分と添付すべき資料 | 独立行政法人 医薬品医療機器総合機構
- [3]第7章 CTD(モジュール3を中心に)承認申請資料の作成