原薬GMPとは何か?ガイドラインの概要や実務上の必須知識をやさしく解説

医薬品の製造には厳格な品質管理が求められます。その中でも原薬(API:Active Pharmaceutical Ingredient)は、医薬品の有効成分そのものであり、その品質は最終製品の安全性や有効性に直結します。
原薬の製造には、特有の品質管理ルールが存在し、それが「原薬GMP」と呼ばれる基準です。
本記事では、「原薬GMPとは何か」を軸に、(製剤の)GMPや治験薬GMPとの比較を交えながら、原薬GMPの基礎知識をわかりやすく解説します。
1.原薬GMPとは
「原薬GMP」は、医薬品の有効成分である原薬を製造する際に一定の品質を保つための管理基準です。
GMP自体は「医薬品の製造管理および品質管理の基準」であり、医薬品全体に共通するルールですが、原薬は製剤とは異なる特性を持つため、原薬専用のGMPが定められています。
[※関連記事:《医薬品製造関係者の常識》GMPって何?|GMPの3原則など必須前提知識をチェック!]
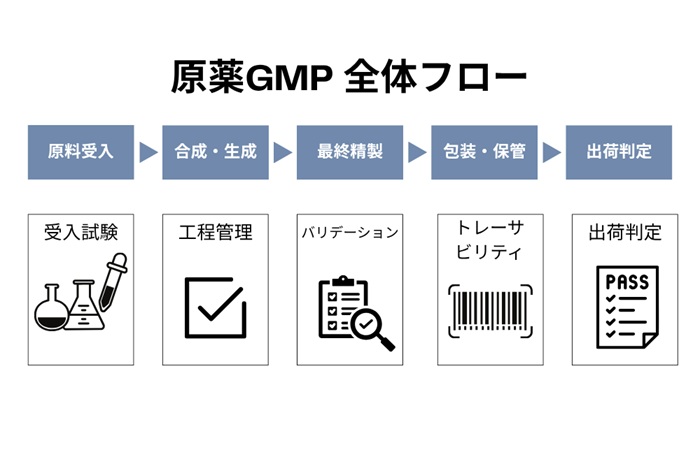
【図1 原薬GMP全体フロー】
原薬GMPは、製品の製造工程だけでなく、原材料の受け入れ、工程管理、試験、保管、出荷に至るまで、全プロセスでの適正管理を求めるものです。また、トレーサビリティの確保や逸脱の管理、変更管理、バリデーションの実施も重要な要素となります。
2.原薬GMP・GMP(製剤)・治験薬GMPの違い
医薬品GMPは「医薬品を適正に製造・管理するための共通言語」ですが、対象やリスク特性によって「原薬GMP」「GMP(製剤)」「治験薬GMP」があります。医薬品GMPの各特徴を表に1示しました。
[※関連記事:治験薬GMPって何?初心者向けにわかりやすく解説!]
【表1 原薬GMP・GMP(製剤)・治験薬GMPの違い】
| 比較項目 | 原薬GMP | GMP(製剤) | 治験薬GMP |
| 適応対象 | 有効成分(API) | 最終製品 (錠剤・注射剤など) |
臨床試験用製品 |
| 変更頻度 | 中程度 (スケールアップあり) |
低い (固定工程が前提) |
高い (処方や工程変更が頻発) |
| 重要な管理ポイント | 供給者監査、工程バリデーション、トレーサビリティ | 無菌性、最終包装、外観検査 | 被験者安全、文書化された変更管理、迅速なリスク評価 |
| 微生物・包装管理 | 比較的緩やか (但し無菌APIは厳格) |
最も厳格 | 被験者リスクを考慮し設定 |
| 代表的なガイドライン |
|
|
|
下図は、3種類のGMPの違いを「製造スケール」「変更の柔軟性」「無菌管理」の三つの視点で整理した早見表です。色の濃淡で要求レベルの強弱を示していますので、全体像が把握できます。

【図2 医薬品GMPの比較】
3.原薬GMPにおけるガイドラインの概要
原薬GMPの運用にあたっては、各国・各地域で定められたガイドラインに基づいて対応します。代表的なものは以下の通りです。
(1)ICH Q7ガイドライン
ICH(International Council for Harmonisation)が発行する「ICH Q7」が、原薬GMPの国際的な標準ガイドラインです。以下の要素が主要なポイントです。
- 原材料の選定と管理
- 製造工程の記録とバリデーション
- 変更管理および逸脱管理
- 試験・保管・出荷の管理
ICH Q7はEUや日本、アメリカなど多くの国で採用されており、グローバルでの原薬製造における標準ルールとされています。
(2)原薬GMPガイドライン(医薬発第1200号)
「原薬GMPガイドライン」(医薬発第1200号、厚生労働省医薬局長通知)は、ICH Q7合意を踏まえて策定された国内指針で、ヒト用医薬品に使用する原薬(API)の製造管理・品質管理の“標準的なあり方”を示しています。ガイドラインは法規上の強制力を持つわけではありませんが、GMP規則等への適合を前提に、原薬の品質と純度を一貫して保証する実務モデルとして位置づけられています
対象範囲は原材料の受入れから製造、包装、保管、出荷に至る全工程で、無菌原薬の場合は滅菌前工程までをカバーします。構成は20章立てで、品質マネジメント、文書化・記録、バリデーション、変更管理、苦情・回収などの共通的要求事項に加え、細胞培養・発酵由来API(第18章)や臨床試験用API(第19章)といった特殊テーマも章立てで詳細に規定しています。
4.原薬GMPの実務的なポイント
(1)原料・資材の供給者管理
原薬製造において最初のステップである原材料の受け入れは、品質の安定性に直結します。供給者を管理し以下の対応が求められます。
- 供給者の評価・監査:
原料や中間体の供給者に対して、事前に品質システムや製造工程の監査を行い、適格性を確認します。重要原料についてはリスクに応じた頻度で定期的な再評価も実施します。 - 供給者との品質契約:
品質不良が生じた際の責任範囲や対応方法を明確にするため、品質に関する技術契約(Quality Agreement)を締結します。 - 受入試験の実施:
ロット毎に原料を試験して適合を確認。場合によってはSkip試験(一定条件下での試験省略)も導入されますが、その科学的根拠と記録が必須です。
(2)製造記録(バッチレコード)の整備とレビュー
製造工程での作業内容を記録する「製造記録(バッチレコード)」は、原薬GMPの中心的文書です。
- 記録の完全性とリアルタイム性:
作業者は”その場でその都度”記録する必要があり、後からまとめて記載することは禁止されています。電子記録の場合も監査証跡(Audit Trail)の確保が求められます。 - レビューの体制:
記録の内容は品質保証部門(QA)によってレビューされ、逸脱や不備がないかを確認したうえで出荷判断が行われます。 - 記録ミスの訂正ルール:
訂正は元の記載が読めるように消し、日付・署名・理由を明示するなど、規定された訂正方法を厳守します。
(3)逸脱管理と変更管理
予期せぬ出来事(逸脱)や製造条件・装置の変更は、製品の品質リスクにつながる可能性があります。
- 逸脱管理:
異常な測定値、設備トラブル、文書ミスなどが発生した際には「逸脱報告書(Deviation Report)」を作成し、原因調査と再発防止策を記録します。重大な逸脱は品質保証部門や上位管理層の承認が必要です。 - 変更管理(Change Control):
原料メーカーの変更、製造パラメータの見直し、分析手順の修正などは、リスク評価(Impact Assessment)を行ったうえで、計画的に進めます。変更は文書化され、影響を最小限に抑える体制が必要です。
(4)工程バリデーションと設備の適格性確認(Qualification)
バリデーションは「その工程で製品を常に意図した品質で製造できること」を立証するための活動です。
- プロセスバリデーション:
製造スケールの再現性や均一性を確認するため、工程の恒常性を実証するのに十分なロット数で統計的に評価します。特に最終精製や結晶化工程は重点項目です。 - 洗浄バリデーション:
製造機器の洗浄が適切に行われていることを確認します。特にマルチ製品ラインではクロスコンタミネーション防止が重要です。 - 設備の適格性確認(DQ/IQ/OQ/PQ):
設計時適格性評価(DQ)、設備据付時適格性評価(IQ)、運転時適格性評価(OQ)、製品製造における性能適格性評価(PQ)を文書化し、承認するプロセスです。
(5)試験・品質管理業務の信頼性確保
原薬の規格試験(純度、含量、不純物など)は、製品品質を直接的に評価するため正確さが極めて重要です。
- 分析法バリデーション:
分析方法の正確性(Accuracy)、再現性(Precision)、直線性(Linearity)などを確認し、バリデーションを行います。 - 安定性試験:
原薬の保存期間と保存条件を決定するための長期安定性試験です。ICH Q1A に準拠し、複数ロットで実施します。
最終的にGMPを実行するのは「人」です。従業員の訓練と意識の醸成は不可欠です。
- 教育訓練の記録:
教育訓練は適任者による定期実施が求められ、内容・実施者・参加記録を管理します。新入社員や異動者には個別の導入教育も必要です。 - 人的エラーの再発防止:
ヒューマンエラーを起こしやすい工程にはチェックリストやダブルチェック体制を導入、標準作業手順書(SOP)の見直しも行います。
これらの実務ポイントを確実に実行することで、原薬の品質と安全性を高い水準で確保できます。GMPは単なるルールではなく、組織全体の品質文化を支える土台でもあるのです。
5.原薬GMPに関する最新動向
(1)ニトロソアミン類の管理について
ニトロソアミン類は発がん性懸念不純物です。
FDAやEMAの通知において、API~最終製剤までのリスク評価と管理が必須となりました。
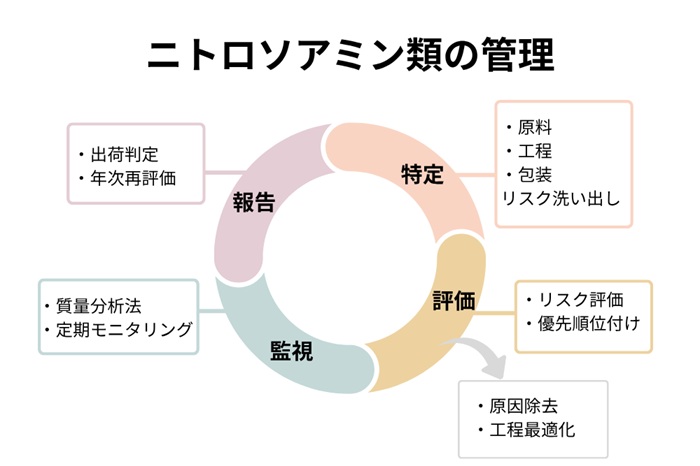
【図3 ニトロソアミン類の管理】
具体的な管理方法は、合成原料・溶媒・工程条件を洗い出し、許容一日摂取量以下に抑える制御戦略を策定、原因除去を最優先とします。ニトロソアミン類はバリデートされた適切な質量分析法で評価し、原薬・製剤・包装由来の生成を定期モニタリングしなければなりません。
ニトロソアミン類が原因で製品の回収も発生しています。原因は原薬製造工程での窒素酸化物(NOx)の管理不足です。原薬段階のニトロソアミン評価が不十分なことが原因で、最終製剤の回収に直結した象徴的な事例となりました。
(2)Annex21への準拠(EU圏に医薬品を輸出する場合)
「Annex 21」は 2022年に公布・発効したEU-GMPの輸入規定です。
日本企業がEU市場へ医薬品を輸出する際は、EU側の輸入者(MIA ホルダー/QP)が「Annex 21」を満たせるよう、EU-GMP適合証明やバッチ記録、試験データを提供する必要があります。
「Annex 21」の主対象は最終製品ですが、QPはサプライチェーン全体のGMP適合性を確認するため、原薬メーカーも監査報告やデータ提供で密接に連携することが求められます。
6.まとめ|セミナー受講のススメ
原薬GMPは、医薬品の基礎を支える最重要な管理基準の一つです。「原薬GMPとは何か」が理解できると、医薬品開発や製造における品質保証の本質を掴めます。また、「GMP」「治験薬GMP」との違いを知ることは、正しい運用と規制対応のためにも不可欠です。
しかし、GMPは文書で理解するだけでなく、実際の事例や運用方法に触れることで初めて「使える知識」となります。もし、これから原薬GMPの業務に携わる予定がある、または今後より深く関わる可能性がある方には、専門のセミナー受講をおすすめします。規制の最新動向や、実際の製造現場での課題と対応策、査察対応のノウハウなど、業務に即した知識が身につきます。GMP対応の向上は、企業全体の信頼性強化にも直結しますので、ぜひ受講してください。
(日本アイアール株式会社 特許調査部 S・O)