医薬品GMPの6つのサブシステムとは?GMP適合性調査での確認ポイントを解説

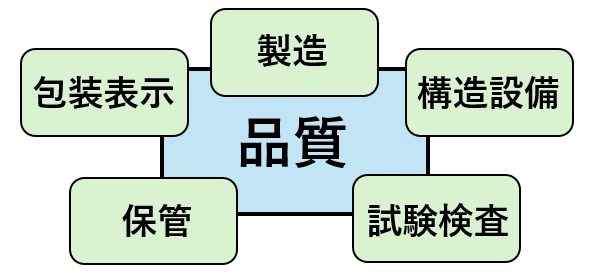
医薬品の品質は、製造工程だけで決まるものではありません。製造設備の管理、試験検査、原材料や製品の保管、包装表示など、製造所のさまざまな仕組みが適切に機能して初めて、安全で品質が確保された医薬品を患者へ届けることができます。
そのため医薬品等の製造所(工場など)がGMPに適合しているか調査する際には、製造所全体を6つの機能的な区分に分けて確認する「6つのサブシステム」という考え方が活用されています。
本記事では、この6つのサブシステムの内容とGMP適合性調査でどのように活用されているのかを解説します。
目次
1.医薬品GMP適合性調査における「6つのサブシステム」とは
医薬品製造において、GMP(Good Manufacturing Practice:医薬品の製造管理及び品質管理の基準)への適合は、品質が確保された医薬品を安定して製造・供給するための重要な前提となります。
医薬品の製造販売承認申請時や承認事項の一部変更時など、製造所のGMP適合性が確認される場面では、製造所がGMPに適合しているかについてGMP適合性調査を受ける必要があります。
[※関連記事:GMPって何?|GMPの3原則など必須前提知識をチェック!]
GMP適合性調査では、製造所全体を体系的に確認するために「6つのサブシステム」という考え方が用いられています。6つのサブシステムは、製造所のGMP活動を、品質、構造設備、製品原料資材保管等、製造、包装表示、試験検査の6領域に区分し、GMP省令の個々の要求事項への適合性に加えて、製造所の管理が全体として効果的に機能しているかを確認するための枠組みです。
【図1 6つのサブシステムの全体像】
【図2 6つのサブシステムのレベル別評価】
また、GMP調査については、厚生労働省から「GMP調査要領」が通知されており、「製造所全体についてのGMP調査においては、表に示す製造管理及び品質管理の主たるサブシステムを踏まえて行うことにより、GMP省令の個々の要求事項への適合性に加え、製造所の管理が効果的に機能しているかを総合的かつ効率的に評価すること。」と記載されています。
以下で、各サブシステムの調査項目について確認していきます。
(※GMP調査要領 表3:医薬品・医薬部外品GMP調査に係るサブシステム を引用)
【表1 医薬品・医薬部外品GMP調査に係るサブシステム】
| サブシステム | 調査項目 | |
| 1.品質 | 1:組織 2:医薬品製品標準書、医薬部外品製品標準書 3:文書管理(データインテグリティ(以下「DI」という。)を含む) 4:出荷管理 5:変更管理 6:逸脱管理 7:品質情報・品質不良(苦情) 8:自己点検 9:回収処理 10:GMP教育訓練 |
11:製造販売業者との合意事項(承認事項を含む。)の遵守 12:品質方針・品質目標 13:品質マニュアル 14:製品品質の照査 15:是正措置・予防措置 16:原料等の供給者管理 17:製造業者等(責任役員)の責任 18:マネジメントレビュー・内部の情報伝達・資源の配分 19:技術移転 20:品質リスクマネジメント 21:外部委託業者管理 |
| 2.構造設備 | 1:手順書・記録書(DIを含む) 2:図面管理 3:建屋・施設(作業室含む)及び設備の適格性確認(製造用水・製造設備・空調設備) 4:設備・機器管理(メンテナンス) 5:コンピュータ化システムの管理 6:校正 7:製造用水管理 |
8:空調管理 9:遮光管理 10:出入り口管理 11:構造躯体管理 12:衛生管理 13:防虫・防そ管理 14:交叉汚染の防止 15:封じ込め措置 |
| 3.製品原料資材保管等 | 1:手順書・記録書(DIを含む) 2:受け入れ管理 3:区分保管管理 4:表示管理 5:出納管理 6:不合格品管理 7:施設及び設備の適格性確認 |
8:設備・機器管理 9:校正 10:衛生管理 11:環境管理 12:防虫・防そ管理 13:出荷作業 14:教育訓練 |
| 4.製造 (1) 一般 (2) 無菌 (3) 生物由来 (4) 放射性 |
(1) 一般 | |
| 1:手順書類 2:製造指図書・記録書管理(DIを含む) 3:作業前確認 4:工程管理 5:異物混入・汚染・混同防止 6:設備・機器管理 7:校正 8:動線 |
9:ゾーニング(区分) 10:防虫・防そ管理 11:作業着管理 12:衛生管理 13:環境管理 14:微生物学的モニタリング 15:バリデーション 16:教育訓練 |
|
| (2) 無菌 | ||
| 1:手順書類 2:製造指図書・記録書管理(DIを含む) 3:作業前確認 4:工程管理 5:異物混入・汚染・混同防止 6:設備・機器管理 7:校正 8:動線 9:ゾーニング(区分) 10:防虫・防そ管理 11:作業着管理 |
12:衛生管理 13:環境管理 14:微生物学的モニタリング 15:バリデーション 16:教育訓練 17:エンドトキシン管理 18:無菌プロセスシミュレーション(Aseptic Process Simulation:APS) 19:清浄化(サニタイズ) 20:浮遊塵埃管理 21:滅菌管理 22:消毒剤等管理 |
|
| (3) 生物由来 | ||
| 1:手順書類 2:製造指図書・記録書管理(DIを含む) 3:作業前確認 4:工程管理 5:異物混入・汚染・混同防止 6:設備・機器管理 7:校正 8:動線 9:ゾーニング(区分) 10:防虫・防そ管理 |
11:作業着管理 12:衛生管理 13:環境管理 14:微生物学的モニタリング 15:バリデーション 16:教育訓練 17:原料入手・取扱い・保管管理 18:ウイルス等の除去・不活化工程の製造管理 |
|
| (4) 放射性 | ||
| 1:手順書類 2:製造指図書・記録書管理(DIを含む) 3:作業前確認 4:工程管理 5:異物混入・汚染・混同防止 6:設備・機器管理 7:校正 8:動線 9:ゾーニング(区分) 10:防虫・防そ管理 |
11:作業着管理 12:衛生管理 13:環境管理 14:微生物学的モニタリング 15:バリデーション 16:教育訓練 17:放射性原料入手・保管管理 18:放射線被爆確認管理 19:放射性物質廃棄管理 |
|
| 5.包装表示 | 1:手順書・記録書(DIを含む) 2:作業前確認 3:表示材料管理 4:工程管理 5:汚染・混同防止 6:施設及び設備の適格性確認 7:設備・機器管理 8:校正 |
9:衛生管理 10:作業着管理 11:動線 12:ゾーニング(区分) 13:防虫・防そ管理 14:環境管理 15:バリデーション 16:教育訓練 |
| 6.試験検査 | 1:手順書・記録書(DIを含む) 2:検体採取・検体管理 3:施設及び設備の管理(試験検査設備・装置の適格性評価・校正並びに試験検査方法の適格性評価) 4:設備・機器管理 5:校正 6:試薬・試液・標準品管理 7:試験用水管理 8:試験動物管理 9:試験検査結果判定・逸脱管理 10:合格ラベル・情報管理(合格情報を保管管理担当者等に伝達する場合等) |
11:参考品・保存品管理 12:衛生管理 13:安定性モニタリング 14:バリデーション(分析法バリデーション) 15:委託試験管理 16:教育訓練 17:試験室環境管理 18:微生物試験管理 19:無菌試験管理 |
(1)品質サブシステム
品質サブシステムは、6つのサブシステムの中核となる領域です。ここでは、組織体制、文書管理、変更管理、逸脱管理、CAPA*1)、教育訓練、自己点検、製品品質照査など、品質保証活動全般が確認されます。
特に近年の調査では、データインテグリティ(DI, Data Integrity)*2)への対応が重要視されており、電子データの帰属性・判読性・同時性・原本性・正確性などを確保するため、アクセス権限管理、監査証跡、バックアップ管理などが適切に実施されているかが確認されます。
[※関連記事:改正GMP省令の重要ポイントを解説!データインテグリティなどの概要がわかる]
*1) 「是正措置(Corrective Action)」と「予防措置(Preventive Action)」の頭文字をとったもので、トラブルの再発防止と未然防止により、品質と安全性を継続的に向上させる仕組みを指します。
*2) データの「完全性(Integrity)」を意味し、データや記録が完全で、一貫しており、正確であること、つまり、データの欠損、不整合、改ざん、捏造などがない状態を意味します。
(2)構造設備サブシステム
構造設備サブシステムでは、建屋、製造設備、空調設備、製造用水設備*3)など、医薬品品質を支えるインフラが適切に維持管理されているかが確認されます。
GMP調査では、設備適格性評価*4)が体系的に実施されているかが重要な確認事項となります。特に無菌製剤では、HVAC(暖房/Heating・換気/Ventilation・空調/Air Conditioning)管理、差圧管理*5)、清浄度管理などが製品品質に直接影響するため、厳格な運用が求められます。
さらに、近年ではコンピュータ化システムバリデーション(CSV, Computerized System Validation)*6)の重要性も高まっています。製造や試験で利用されるシステムが適切に管理され、データ改ざんなどのリスクが低減されているかも重点的に確認されます。
*3) 医薬品の製造、原料の溶解、製造設備の洗浄や滅菌などに用いられる水のことで、製品品質に影響を与えないよう、不純物や微生物を適切に管理し、厳格な品質基準を満たさなければなりません。
*4) 製造設備が「適切に設計・据え付けられ、仕様通りに正しく作動し、安定した結果を出し続けること」を客観的に検証し、文書化することです。設計時適格性評価(DQ)、据付時適格性評価(IQ)、運転時適格性評価(OQ)、性能適格性評価(PQ)の4つの段階(4つのQ)に分けて評価されます。
*5) 部屋同士の圧力差をコントロールし、空気の流れる方向を制御することで、汚染物質やホコリなどの侵入を防止したり(陽圧管理)、室内の汚染された空気が他の部屋に漏れ出すことを防ぎ(陰圧管理)ます。
*6) バリデーション(Validation)とは、「検証」「妥当性確認」を意味する言葉で、製造等のプロセスが「意図した品質の製品を、常に一貫して安全に製造できる状態であるか(この器具・設備を使い、この手順で作業すれば、誰がいつ製造しても同等の品質のものができあがる(同等の結果が出る)こと)」を科学的に検証・証明し、文書として記録することです。
コンピュータ化システムバリデーションでは、コンピュータ(ソフトウェア等)で制御しているシステムについて、あらかじめ定められた要求仕様どおりに動作し、一貫して期待した結果を得られることを検証し、文書化します。
(3)(製品原料資材)保管等サブシステム
原料や資材の管理は、製品品質を確保するための出発点です。製品原料資材保管等サブシステムでは、受入試験*7)、区分保管*8)、表示管理(例:原料のステータス表示として「不合格品」のラベルを付与する)、不合格品管理*9)、在庫管理などが確認対象となります。
例えば、原料の取り違えや誤出庫が発生すれば、重大な品質問題につながる可能性があります。そのため、識別表示、ロット管理、先入れ先出し(FIFO:First In, First Out)、温湿度管理などが適切に行われているかが重要です。
なお、原料等の供給者管理は主に品質サブシステムの調査項目に含まれますが、受け入れ管理や保管管理とも密接に関係します。供給者監査や品質取決めを通じて、原料供給段階から品質を確保する考え方が求められます。
*7) 製造に使用する原料や資材が、あらかじめ定められた品質規格に適合しているかを受け入れた時に確認する検査
*8) 医薬品などを取り扱う現場において、品質の劣化や誤使用、異物混入、交叉汚染を防ぐために、状態や種類ごとにエリアを分けて管理すること
*9) 仕様や品質基準を満たさない製品や原材料を明確に識別し、隔離・処分することで市場への流出や製品への混入を防ぐこと
(4)製造サブシステム
製造サブシステムでは、実際の製造工程がGMPに従って運用されているかを確認します。製造指図記録書*10)、工程管理、ラインクリアランス*11)、交叉汚染防止*12)、バリデーションなどが主な調査対象となります。
特に無菌製剤では、無菌操作、環境モニタリング*13)、APS(Aseptic Process Simulation、無菌プロセスシミュレーション)、滅菌管理などの高度な管理が要求されます。さらに、生物由来製品*14)ではウイルス不活化工程など、放射性医薬品では放射線管理など、製品特性に応じた管理も必要です。
製造現場では「記録に残っていないことは、実施していないとみなされる」という考え方が基本であり、記録の完全性を確保することが極めて重要です。
*10) 製品の製造手順や条件を指示する「製造指図書」と、実際の作業実績や測定結果を記入する「製造記録書」を一体化した文書
*11) 作業エリアや製造・包装ライン上に、前回の製造工程で使用した製品、原料、資材、文書などが一切残っていないこと(クリアランス)を確認する手順
*12) 交叉汚染(交差汚染)とは、製造環境において原料や製品に、本来含まれるべきではない他の成分や物質が混入・付着することです。防止策として、前回使った設備の洗浄、異なる原料などを扱う作業を同じ部屋で行わない(作業室の専用化)などがあります。
*13) 空気中の浮遊微粒子や微生物(浮遊菌)、落下菌などを継続的に監視・記録し、汚染を防ぐこと
*14) 人やその他の生物(植物を除く)に由来する細胞・組織などを原材料として製造される医薬品・医療機器等
(5)包装表示サブシステム
包装表示は、患者への最終的な情報提供にも関わる重要工程です。この包装表示サブシステムでは、表示材料管理*15)、包装ライン管理、誤表示防止、ラインクリアランスなどが確認されます。
医薬品の取り違えや表示ミスは、患者の健康被害につながる可能性があります。そのため、バーコード照合、電子照合システム、ダブルチェックなど、多重の防止策が求められています。
また、近年ではシリアライゼーション(Serialization)*16)や偽造防止対策への対応も重要性が高まっています。
*15) 表示材料、つまり、製品等について表示する資材(ラベル、添付文書、個装箱等)そのものについての管理です。例えば、法改正などにより表示材料に記載しなければならない事項が追加された際には、新版のラベル等に置き換える必要があります。
*16) 医薬品の包装1つひとつに固有のシリアル番号(個体識別コード)を付与し、製造から流通、消費までのサプライチェーン全体で個々の医薬品を追跡できるようにする仕組み
(6)試験検査サブシステム
試験検査サブシステムでは、原料、中間製品、最終製品などに対する品質試験が適切に実施されているかが確認されます。試験法バリデーション、分析機器管理、標準品管理、安定性試験*17)、OOS(Out of Specification、規格外試験結果)*18)対応などが主な確認項目です。
分析データの信頼性は医薬品品質保証の根幹であり、近年ではデータインテグリティへの要求が高まっています。
例えば、根拠や手順が不明確なクロマトグラムの再解析、都合のよい結果の選択、未記録データの存在、共有IDの使用、監査証跡の確認不足などは、重大な指摘につながる可能性があります。
また、微生物試験や無菌試験では、試験環境そのものの管理も重要であり、清浄度の管理や作業者教育が適切に実施されているかも確認されます。
*17) 医薬品などの品質が時間経過とともにどのように変化するか(または変化しないか)を確認する試験
*18) 原材料や製品の試験・検査において、あらかじめ設定された基準値や合格範囲から外れた測定結果が出たこと
2.おわりに
GMP適合性調査における6つのサブシステムは、単なるチェックリストではなく、製造所全体の品質システムが有効に機能しているかを多面的に評価するための枠組みです。
近年は、PIC/S GMPガイドラインなど国際的なGMP水準やデータインテグリティへの対応が求められ、調査ではより実効性のある管理状況が確認されるようになっています。
特に重要なのは、各サブシステムが独立して存在するのではなく、相互に連携しながら品質保証体制を形成している点です。品質サブシステムを中心に、構造設備、製造、試験検査、包装表示、製品原料資材保管等の各機能が有機的に結び付くことで、安定した医薬品品質の確保につながります。
今後のGMP対応では、「調査対応のため」ではなく、「患者や使用者の安全を守るため」という本質的な視点がより重要になります。6つのサブシステムを正しく理解し、自社の品質システムを継続的に改善していくことが、信頼される医薬品の製造につながります。
(日本アイアール株式会社 Y・A[薬剤師])